La Bêta Thalassémie est une maladie génétique de l’hémoglobine transmise par les parents.
 Dans les formes sévères (majeure et intermédiaire), la bêta-thalassémie entraine une anémie liée au manque de globules rouges et d’hémoglobine.
Dans les formes sévères (majeure et intermédiaire), la bêta-thalassémie entraine une anémie liée au manque de globules rouges et d’hémoglobine.
Les traitements demeurent lourds et contraignants pour les malades touchés par ces formes; des transfusion sanguines régulières et systématiques sont nécessaires. Elles doivent être accompagnées de la prise quotidienne d’un traitement permettant d’enlever le fer en excès dans l’organisme apporté par les transfusions sanguines car il empêche à terme les organes de fonctionner.
A ce jour, seule la greffe de moelle osseuse peut guérir les malades, mais ce traitement n’est réservé qu’à un petit nombre de malades bénéficiant d’un donneur apparenté.
Depuis quelques années, la recherche médicale avance à grands pas!
La thérapie génique
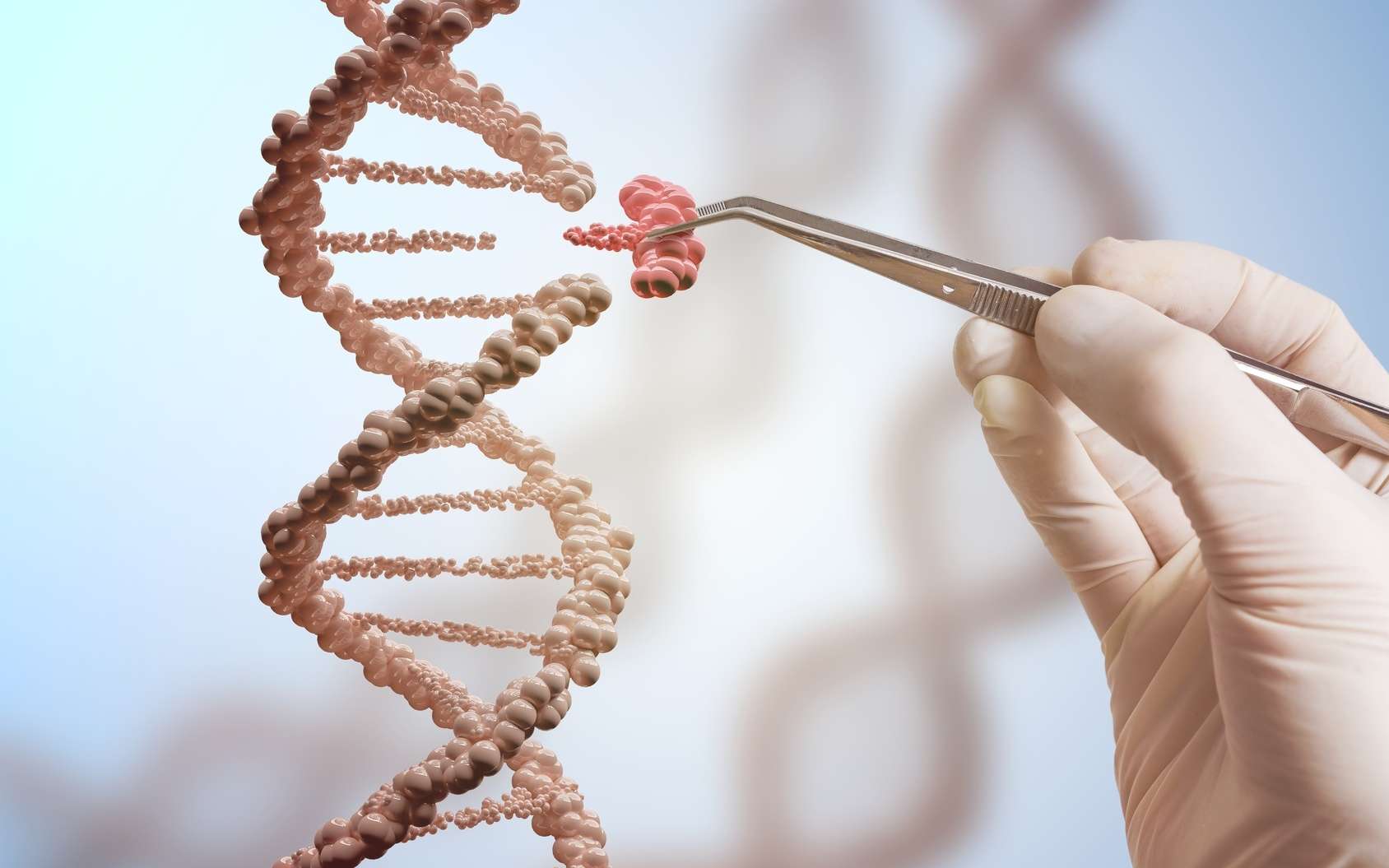
La thérapie génique est un traitement qui permet à l’organisme de produire des globules rouges et de l’hémoglobine en quantité suffisante et ainsi de réduire totalement ou en partie les transfusions sanguines.
- En quoi cela consiste?
Lors du traitement de thérapie génique, les cellules souches du patients sont prélevées. Ensuite, le patient doit subir une chimiothérapie afin que ses cellules souches restantes porteuses de l’anomalie génétique soient détruites.
Les cellules souches qui ont été prélevées sont traitées en laboratoire. On y introduit un gêne thérapeutique qui a la capacité de permettre la production d’hémoglobine.
Les cellules ainsi corrigées sont réinjectées au patient et vont « coloniser » les globules rouges de l’organisme.
Une trentaine de patients ont déjà bénéficié de ce traitement et les résultats sont prometteurs;
Pour ces patients, la thérapie génique a permis soit d’arrêter les transfusions, soit de diminuer la fréquence et la quantité des transfusions;
Ce traitement devrait bientôt être approuvé aux Etats Unis et en Europe, ce qui permettrait de le rendre disponible à un grand nombre de malades.
Luspatercept, protéine de fusion du récepteur de l’activine
Le Luspatercept augmente la production d’hémoglobine en agissant sur le processus de fabrication des globules rouges (erythropoïèse).
- En quoi cela consiste?
Le Luspatercept est un traitement expérimental qui s’administre par voie sous cutanée toute les 3 semaines.

Les résultats des études démontrent une réduction des besoins transfusionnels chez les patients dépendant des transfusions et une augmentation du taux d’hémoglobine chez les patients non dépendant des transfusions.
L’essai clinique va se poursuivre encore pendant 5 ans.
Pour en savoir plus!
La Bêta Thalassémie, fiche Orphanet
La promesse de nouvelles thérapies pour les syndromes bêta thalassémique, article scientifique en anglais
Article rédigé le 18 janvier 2019